Condensation polymerisation: Phenolic (PF) resins. Epoxy (Ep) resins. Polyurethane (PU) resins.
PowerPoint
papers
Index

| Composites Design and Manufacture (Plymouth University teaching support materials) Condensation polymerisation: Phenolic (PF) resins. Epoxy (Ep) resins. Polyurethane (PU) resins. |
Lecture PowerPoint |
Review papers |
Subject Index |
|
Fibre reinforced plastics may be formed using either thermosetting resins or thermoplastic matrix systems. Currently most commercial composites are manufactured by bringing together the reinforcement fibres and a liquid resin then causing the resin to harden using chemicals and heat. The hardening (cure) of the resin is achieved by the formation of a network of cross-links (permanent covalent bonds) between the chains of the polymer.
The choice of a thermosetting resin system is dictated by many parameters (cost, processability and properties). A brief guide to the relative merits of five important polymers of significance for advanced composites is given in Table 1.
| Polymer system | phenol-formaldehyde | epoxides | unsaturated polyesters | methacrylics | polyurethanes | bismaleimides |
| Also known as: | phenolics | generally diglycidyl ethers | UP or UPE | including vinyl esters (VE) | PU | BMI |
| Cost | £ | £££ | £ | £ | £ | £££££ |
| Process temperature | ☀☀☀ | ☀☀ | ☀ | ☀ | ☀ | ☀☀☀☀ |
| Cure temperature | ☀☀ | ☀☀☀ | ☀ | ☀ | ☀ | ☀☀☀☀ |
| Cure time | ºº | ººº | ºº | ºº | º | ººº |
| Use temperature | ☀☀☀ | ☀☀ | ☀ | ☀☀ | ☀ | ☀☀☀ |
| Mechanical properties | ✓ | ✓✓✓ | ✓ | ✓✓ | ✓ | ✓✓✓✓ |
| Environmental resistance | ✓ | ✓✓✓ | ✓✓ | ✓✓ | ✓ | ✓✓✓✓ |
| Flame, smoke and toxicity (FST) | ✓✓✓ | ✓ | ✓ | ✓ | ✓ | ✓✓✓ |
| Surface finish | ✓ | ✓✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ |
A major factor in the selection of the resin is the ability to work with that resin until the component has been formed, and then to cure the component in a reasonable time to realise the required properties. For most thermosetting systems, chemicals are added to the resins to catalyse (and accelerate) the cure. Unless these chemicals are only activated at an elevated temperature, the cure commences as soon as the materials are mixed.
An increase in temperature generally reduces the viscosity and hence eases the flow. However, it also increases the rate of cross-linking. At low temperatures the effect of temperature on viscosity predominates and the total flow occurring before the gel-point (setting to a rubbery state) increases with temperature. At higher temperatures, a further increase in temperature will increase the cure rate and reduce the total flow. The net result is indicated in the Figure below.
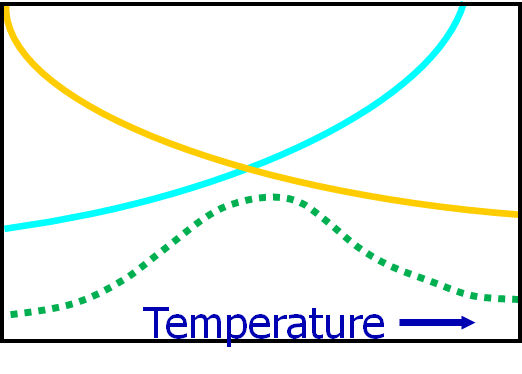
Figure 1: Instantaneous flow is dependent on viscosity (blue line) and on cure (orange line) both as a function of temperature, so the net effect is total flow as indicated by the green
dotted line.
It is important to form the component before cross-linking progresses so far as to prevent flow of the resin. In heated moulds, the low thermal conductivity of the polymer can produce temperature gradients in the resin, and hence both the viscosity and cure rate can vary with both time and position. Also, the viscosity in closed moulds may vary with the flow shear rate at different positions within the mould. Frictional heat from shear will also vary with position, generally being higher close to the cavity wall. In a correctly optimised process the viscosity need not change significantly during forming of the component, as this can be achieved before the onset of major network formation.
It is seldom possible to regenerate useful polymers as the conditions required to break the cross-links will also damage the backbone chain. Such fugitive cross-links are possible for other applications by:
Cross-linked polymers can be divided into two groups according to the cross-link density. The lightly cross-linked polymers (eg: vulcanised rubber) are cured to prevent the material deforming indefinitely under load. The chains can no longer slide past each other and 'flow' is not possible without breaking covalent bonds. The molecular segments between the cross-links remain flexible and the material may exhibit either brittle or rubbery response dependent on the temperature. Crystallisation may be possible both in the unstressed and the stressed state.
In highly cross-linked polymers, the cross-links are closer together and a stiffer network is produced. As the degree of cross-linking increases a tighter less-flexible network results. Segmental motion is restricted and the transition temperature approaches the decomposition temperature. At high cross-link densities only the amorphous rigid (glass-like) state exists. This is typical of epoxy, phenolic and aminoplastic resins.
Resin shrinkage [1]
Many processes involve the use of elevated temperatures. In common with most materials, the polymer will shrink on cooling due to the coefficient of thermal expansion. For perfectly isotropic shrinkage, the total volumetric shrinkage rate of a material, Sv, can be related to the linear shrinkage rate Sl by the Equation:
Sl = 1-(1-Sv)1/3This is often simplified by neglecting higher order terms in the binomial expansion to give Sl ≈ Sv/3. Changes in volume may also occur due to loss of volatile components and/or crystallisation. For thermoplastic in-mould polymerisation systems, changes may arise due to the chemical reaction. For thermosetting systems, changes are due to the cross-linking (cure) reactions with typical values shown in Table 2.
| System | Shrinkage (%) | Source |
| Robnor PX439N epoxy encapsulating resin | 0.3 (volume) | Robnor Resins Technical Data Sheet |
| Gurit PrimeTM 20LV infusion epoxy/extra slow hardener | 1.5 (linear) | Gurit manufacturers product data sheet |
| Gurit PrimeTM 20LV infusion epoxy/fast hardener | 1.8 (linear) | Gurit manufacturers product data sheet |
| Crystic 491/491PA thixotropic isophthalic polyester resin | 2.6 (linear) 7.5 (volume) |
Scott Bader Technical Leaflet TL164. July 1987. |
| Crystic 474PA thixotropic orthophthalic polyester resin (thixotropic pre-accelerated Crystic 198) |
2.8 linear) 8.2 (volume) |
Scott Bader Technical Leaflet TL274, May 2000. |
| Crystic 198 medium viscosity orthophthalic polyester resin | 2.8 (linear) 8.2 (volume) |
Scott Bader Technical Leaflet TL273, May 2000. |
The contractions arising from cure can result in thermal residual stresses, and hence distortion (springback or springforward), surface defects (e.g. sink marks) on the component or in extreme cases matrix cracking.
Cure of resins
Three different mechanisms are involved in cross-linking: condensation, ring-opening and addition reactions. It is convenient to produce relatively low molecular weight structures (A-stage resins) which are comparatively stable at room temperature. These small branched units may then be formed to shape and cross-linked to form the 3-D network (C-stage resin) by catalysis or heat. The stages are:
The cure of a composite progresses towards a position where 100% of all potentially reactive chemical groups have been consumed. However, as this situation is approached the movement of segments of the polymer chain is increasingly constrained and hence the final reactions may not be possible unless both reactive sites get close enough for this to happen. Incomplete cure will limit the shrinkage and hence the resin density achieved and in consequence there will be some additional free volume remaining in the matrix. The incomplete cure will cause the glass transition temperature to be lower than for the fully cured resin. The lower density will mean fewer bonds/m3 and hence lower moduli and strengths (for ambient temperature cure of unsaturated systems, the resin may only reach 80-90% of the mechanical properties obtained at full cure). The additional free volume will permit easier diffusion of chemicals and potentially reduce the durability of the resin system. For high-performance composites, it is normal to subject the component to a higher temperature (post-cure) to achieve optimum properties. This will normally be undertaken shortly after production as the unreacted sites on the polymer may become inactive over time.
Phenolic resins were the first truly synthetic resins to be exploited. Butlerov (1859) first described formaldehyde polymers. Adolf Bayer (1872) reported that phenols and aldehydes could be reacted to form resinous substances. Arthur Smith (1879) took the first British Patent (16274) for phenol-aldehyde resins as an ebonite substitute in electrical insulation. Baekeland (1907) discovered techniques of controlling and modifying the reaction to produce useful products. Phenol-formaldehyde materials quickly became established in several fields. Recently their use for fibre composite materials has been promoted by the good FST (flame, smoke and toxicity) properties.
Phenolic resins can be broadly divided into three groups:
Methylolation
The first step in the reaction is the formation of addition compounds by reaction of the formaldehyde (CH2O) at ortho- or para positions on the phenol molecule (C6H5OH, represented by Φ below, including in cases where a hydrogen has been substituted by another group). This is known as methylolation. The products formed may be considered as monomers for subsequent polymerisation. They are most satisfactorily formed under neutral or alkaline conditions.
Φ + CH2O ⇒ Φ CH2OH
Φ CH2OH + CH2O ⇒ Φ (CH2OH)2
Φ (CH2OH)2 + CH2O ⇒ Φ (CH2OH)3
Resoles
In the presence of alkaline catalysts and a molar excess of formaldehyde the methylol phenols can condense either through methylol linkages or through ether linkages. In the latter case, methylene bridge formation may result in the release of water:
HO~CH2~Φ~CH2~OH + HO~CH2~Φ ⇒ HO~CH2~Φ~CH2~O~CH2~Φ or HO~CH2~Φ~CH2~Φ~CH2~OH
The resulting resoles are soluble and fusible and contain alcohol groups. Continued reaction can lead to network formation, although the first product may lose formaldehyde:
HO~CH2~Φ~CH2~O~CH2~Φ ⇒ HO~CH2~Φ~CH2~Φ + CH2O
Resoles are generally neutralised before cure is carried out. Network polymers are then obtained simply by heating. The methylene bridge is effectively formed by the elimination of formaldehyde from the methylol group.
Novolacs
In the presence of acid catalysts and a molar excess of phenol, the methylol derivatives condense with phenol to form dihydroxydiphenylmethane:
Φ~CH2~OH + Φ ⇒ Φ~CH2~Φ + H2O
Further condensation and methylene bridge formation results in fusible and soluble linear low polymers called novolacs:
~Φ~CH2~Φ~CH2~Φ~CH2~Φ~CH2~Φ~
Ortho- and para- links occur at random. Molecular weights may be as high as 1000, corresponding to around 10 phenyl groups. These materials do not react further to give cross-linked resins, unless the phenol:formaldehyde ratio is reduced below 1.
Conversion of novolacs into network polymers can only be accomplished after addition of a cross-linking agent. This may be paraform or further formaldehyde, although HMT [hexamethylenetetramine: (CH2)6N4] is almost invariably used:
2 (CH3)2Φ ⇒ (CH3)2ΦCH2~NH~CH2~Φ(CH3)2
3 (CH3)2Φ ⇒ [(CH3)2ΦCH2~]3N
When these benzylamines are heated at 180-190°C in the presence of phenol, ammonia or methylamine are evolved:
(CH3)2ΦCH2~NH~CH2~Φ(CH3)2 ⇒ (CH3)2Φ~CH2~Φ(CH3)2 + NH3
[(CH3)2ΦCH2~]3N ⇒ (CH3)2Φ~CH2~Φ(CH3)2 + CH3NH2
Extent of cure
In phenolic resins, the network is essentially produced by a condensation reaction (the elimination of small molecules, eg: water). There is evidence to suggest that actual cross-linking may only involve a small fraction of the potential sites.
See also Hubert Monteiro's Introduction to Epoxies
Epoxy resins cure by a ring-opening mechanism. They can be considered as a condensation polymer in which a water molecule has been removed from two adjacent carbon atoms on the main chain. The epoxy group is a three-membered ring containing one oxygen and two carbon atoms (represented by CH2(O)CH~, with bonds on the backbone of the chain represented by ~ and Rx being a further organic group where x is used to discriminate between the groups). The curing mechanisms may be quite complex. When a methylene group separates the epoxy ring from an ether linkage, the ring may be readily attacked by an active hydrogen, active ions or even tertiary amines to form a negatively charged oxygen ion. This ion may simultaneously open up another epoxy group forming a covalent bond and generating a further ion:
R1N + CH2(O)CHR2 ⇒ R1N+~CH2~CHR2O-
R1N + ~CH2~CHR2O- + CH2(O)CHR3 ⇒ R1N+~CH2~CHR2~O~CHR3O-
When the reaction occurs at both ends of the molecule (eg: in diglycidyl ether resins), a cross-linked structure is built-up. The epoxy group, particularly when catalysed, will also react to form an ether link and another hydroxyl group if hydroxyl groups are present:
R1~OH + CH2(O)CHR2 ⇒ R1~O~CH2~CH(OH)~R2 or H~O~CH2~CH(OR1)~R2
R1~CH2~CH(OH)~R2 + CH2(O)CHR3 ⇒ R1~CH2~CHR2~O~CH2~CH(OH)~R3
In addition to the catalytic reactions, the resin may be cross-linked by agents which bridge two molecules either at the epoxy ring or at the hydroxyl groups. For example, with a primary amine the two hydrogen atoms will form hydroxyl groups with the epoxide ring and an amine link with each second carbon atom:
R1CH(O)CH2 + H2NR2 + CH2(O)CHR3 ⇒ R1CH(OH)~CH2~NR2~CH2~CH(OH)~R3
With a dicarboxylic acid, two hydroxyl and two ester groups are formed:
R1CH(O)CH2 + HOOC~R2~COOH + CH2(O)CHR3 ⇒ R1CH(OH)~CH2~OOC~R2~COO~CH2~CH(OH)~R3
The above reactions can only lead to chain extensions, but polyamines (with more than two active hydrogen atoms) can lead to cross-linking. Acids and anhydrides may react with hydroxyl groups leading to chain branching by true condensation (elimination of water):
R1~COOH + HO~R2 ⇒ R1~COO~R2 + H2O
Typical epoxy laminating systems
Resin selection involves compromise between manufacturing considerations and the service environment of the component. Large components will need a long pot-life (time in a "usable" state) during fabrication. However cold curing resins tend to have lower glass transition temperatures and poor moisture resistance in comparison with hot cure resins. In cold- cure resins it is particularly important to use small batches as a large mass can produce an exothermic reaction which reduces the pot life. Further considerations which may affect the manufacturing include the time for the laminate to reach handling strength, and the allergic nature of the resin components.
The "usable" pot life of a resin is usually quoted with reference to a viscosity at that time, though this may be somewhat arbitrarily chosen. For Araldite system resins the pot-life is quoted as the time from mixing to reaching 15 poise (1500 mPas) viscosity. At this value the ability to laminate is reduced because:
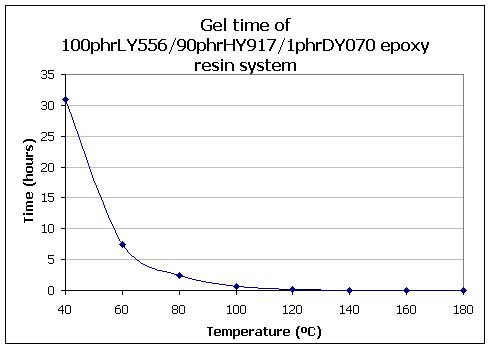
Epoxy laminating systems can be considered in two categories: cold/warm curing (up to 60°C), and hot curing resins. Typically a hot curing resin will gel in 7-8 hours at 60°C, or in 10-12 minutes at 120°C. The variation of gel time with temperature is shown in the Figure below for the LY556/HY917/ DY070 resin system when used pure in small quantities. Fibre content and laminate thickness may modify the gel time considerably.
The resin system MY750/HY917/1%DY070 is suitable for laminating in reasonably warm conditions. The initial viscosity at 25°C is under 10 poise, and varies with time and temperature as indicated in the Figure below. However in a cold environment it will be necessary to warm the resin pot to around 35°C to reduce the viscosity.
A resin system such as XD893/HY932 is unsuitable for room temperature laminating because the initial viscosity is 30-90 poise. It requires warming to 50°C. It is labelled as specially suitable for hand lay-up.
The resin matrices for pre-impregnated (prepreg) materials are different from those employed in hand laminating as there is a requirement for the material to be taken to, stored, and subsequently handled and laid-up in the B-stage (partially cured). Long shelf lives (typically 12-18 months at -18°C) and reasonable 'out times' (usable life at room temperature for cutting and lay-up, perhaps 2-3 months) are required. These factors lead to prepreg systems usually having hot-cure matrix resins with minimum cure temperatures around 120°C. Lower temperature prepreg systems are available, primarily for the manufacture of composite tools. They have (predictably) reduced shelf life.
Importance of resin viscosity in composite manufacture
Stringer [1] has studied the optimization of the wet lay-up/vacuum bag process, for the production of composites with high fibre volume fraction and low void content. It is widely considered that wet lay-up composites fabricated in a vacuum bag suffer from an inherently high void content compared to autoclaved prepreg components. The void content in vacuum bagged carbon fibre reinforced epoxy resin can reach or exceed 10%. The most probable cause is that vacuum bag consolidation is commonly used without consideration of the changing resin viscosity. Stringer has proposed that the wet lay-up technique can be improved by incorporating a dwell period at the start of the cure cycle to increase the resin viscosity before applying the bagging pressure. Resin viscosity is the critical parameter in producing fibre composites with low void content.
A dwell time 'window' exists for each resin system within which high quality laminates can be consistently produced with up to 58 v/o fibre and less than 2% voidage. This window exists between the same viscosity limits regardless of the resin system and temperature being used and can thus be determined from the viscosity/temperature/time characteristics for the resin under consideration.
Stringer [2] concludes that:
Recyclamine® (formerly Connora Technologies, now Aditya Birla Chemicals, Mumbai ~ India) technology is a patented novel amine-based curing agent system with specifically engineered cleavage points at cross-linking sites. Cleavage converts the thermosetting epoxy into a thermoplastic under specific conditions. The system enables recycling of epoxy thermoset composites, thus contributing towards a circular economy.
CompositesWorld article 31-Nov-2014). CompositesWorld article 08-Dec-2016. IBEX flyer.
L&L Products Inc (Romeo MI ~ United States of America) markets an advanced engineering thermoplastic adhesive hybrid resin, T-Link™, that is claimed to combine "superior adhesion with the processing characteristics of a thermoplastic". The material is available as powder, pellet, film or fibre, and does not require refrigerated storage. It is a "true epoxy [which] at 135°C turns itself into a thermoplastic" [Eric Taylor, Carr Reinforcements, email dated 29-Mar-2022].
Nautilus Composites LLC provide a single source for polyurethane composite technology, including:
References
Further reading